VASP 使用
VASP 使用
介绍
- VASP 全称:Vienna Ab-initio Simulation Package
VASP ELFCAR 文件
VASP6 用的赝势和 5.4.4 相同
Perdew-Burke-Ernzerhof (PBE) 形式的 generalized gradient approximation (GGA) 泛函(表达电子间的交换关联作用)
投影缀加平面波赝势(PAW)方法(描述离子 - 电子相互作用)
能带计算,ISMEAR=0?
EIGENVAL 文件内容含义
DFT-D3:vdW 相互作用修正
Heyd–Scuseria–Ernzerhof 泛函 (HSE06):更精确,处理电子和光学性质
+U
1
2
3
4
# LDAU 等参数
LDAU
LDAUU
LDAUJ
自洽计算:均匀 K 点计算(严格一点,先弛豫后静态计算;模型较好,不 care 晶格常数,可直接静态计算) 非自洽计算:特殊 K 点
METAGGA
SCAN (Strongly constrained and appropriately normed)
参考资料
- VASP INCAR 参数:Category:INCAR tag - Vaspwiki
- VASP POSCAR:POSCAR - Vaspwiki
- VASP KPOINTS:KPOINTS - Vaspwiki
- VASP 赝势推荐:Available PAW potentials - Vaspwiki
- VASP 输出文件:Category:Output files - Vaspwiki
- VASP Manual:The VASP Manual - Vaspwiki
- VASP Categories:Categories - Vaspwiki
- VASP Tutorial:Category:Tutorials - Vaspwiki、Tutorials
- VASP Examples:Category:Examples - Vaspwiki
| [VASP中POTCAR使用指南 | Jun’s Blog](https://next.jun997.xyz/2022/04/14/ba8ff0b84c20.html) |
输入文件及参数介绍
VASP 中计算电子基态的算法
Algorithms used in VASP to calculate the electronic groundstate - Vaspwiki
k 点积分
phonon dispersion 计算
原子计算
分子动力学计算
GW 计算
内含 VASP 计算相关的案例
GitHub - hello-arun/tutorials: Tutorials of codes such as VASP, Quantum Espresso and Lammps
VASP 计算流程:VASP的计算流程 - Jun’s Blog
- NCORE: 指定单个轨道计算所使用的核数量
- NPAR: 指定同时并行处理的能带数
- KPAR: 指定同时并行处理的 K 点数量
使用
工具
VASPMO:用于显示 VASP 计算的波函(或分子轨道)。它能够读取 VASP 的输出文件 PROCAR 和 CONTCAR,并产生 Gaussian 输出格式的输出文件,用于其它显示工具,如 Molekel、Chemcraft、Gabedit、Molden 和 JMol 等)读取,进而绘制和观看体系的分子轨道
算例
VASP 官网案例——Calculate U for LSDA+U(线性响应方法求 U 值)
内含表面能、层间距变化计算公式:Ni 100 surface relaxation - VASP Wiki
石墨堆叠方向层间距确定:
- GGA level 的半局域(semilocal)DFT 低估了长程色散相互作用,导致石墨晶格在堆叠方向上的错误高估:8.84Å(PBE)对 6.71Å(exp)。
- 使用 Tchatchenko and Scheffler 方法(添加 IVDW 和 LVDW_EWALD 参数)考虑范德华力相互作用(van der Waals interactions)进行纠正
NiO:反铁磁
Ni(100) 表面的 DOS 计算,没有先进行 SCF 计算? Ni(100) 表面的能带结构计算,K-path 是 reziprok 方式,非 Line-Mode,vaspkit 和 pymatgen 无法获取数据,只能使用 p4vasp?
Ni(111) 表面高精度单点能计算(截断能提高;用以计算吸附能、功函数(添加 LVHAR 参数)):Ni 111 surface high precision - VASP Wiki
[Ex49 功函数(work function)的计算(一) Learn VASP The Hard Way](https://www.bigbrosci.com/2018/09/03/ex49/)
VASP wiki 中的示例 POSCAR 格式和 POTCAR 文件(PAW 格式)较老?
Partial DOS of CO on Ni 111 surface - VASP Wiki
VASP 官网算例中的部分 POSCAR 文件中没有元素符号行(第 6 行,不影响)
- 振动频率计算的意义?NFREE 参数,振动 mode?
1
2
3
4
# 用的是哪个泛函?
TITEL = PAW Ni
TITEL = PAW C
TITEL = PAW O
- K 点网格某方向数值为奇数,$\Gamma$ 中心?
Si 熔化 AIMD 计算:Liquid Si - Standard MD - VASP Wiki
只有 1 个 K 点,可以用 vasp_gam 来运行,加快运行速度
Pair correlation function 数据保存在 PCDAT 文件中
Si 结晶 AIMD 计算(扩散系数及 PCF):Liquid Si - Freezing - VASP Wiki
1
2
3
4
5
6
7
8
#!/bin/bash
for i in 800 900 1000 1100 1200 1300 1400 1500 1600 1700 1800 1900 2000; do
# awk 的作用是什么
awk <PCDAT.$i >pair.$i ' NR==8 {pcskal=$1} NR==9 {pcfein=$1} NR>=10036 {line=line+1; print (line-0.5)*pcfein/pcskal,$1} '
done
gnuplot -e "set terminal jpeg; set key left; set xlabel 'r (Ang)'; set ylabel 'PCF'; set style data lines; plot 'pair.2000','pair.1400','pair.800' " > pair.jpg
- DOS 计算过程中 ISMEAR=0 和 -5 的差别是什么 Part 2: More silicon
输入文件
POSCAR
构型文件,需至少包含体系的几何信息(晶格常数、基矢、元素种类及其数目)和原子位置(以及分子动力学计算时原子的初始速度(不常用));生成 POSCAR 文件是 VASP 计算的起点;可以手动生成,也可以从一些在线晶体学数据库(Material Project、aflow、icsd 等)中获取。
第二行值如果为负数,表示体积
例子:
1
2
3
4
5
6
7
8
9
10
Cubic BN
3.57
0.0 0.5 0.5
0.5 0.0 0.5
0.5 0.5 0.0
B N
1 1
Direct
0.00 0.00 0.00
0.25 0.25 0.25
- 第 1 行:注释行(Comment line);可以对体系进行描述,也可以空着
- 第 2-5 行:缩放因子和基矢(Scaling factor and lattice);只要与体系的晶格常数符合即可
- 第 6-7 行:元素种类(optional,这行可以没有,VASP4 版本没有)及对应数目(Ion species and numbers);元素种类的顺序需与 POTCAR 文件中的一致;
- 第 8-N 行:原子坐标信息(Ion positions);Direct(首字母大小写以及只写 D 均可)表示分数坐标,Cartesian(同上)表示笛卡尔坐标(如果第 8 行是 Selective Dynamics,原子位置后面每个方向需添加 T/F,表示是否对 x y z 方向进行固定)
- 原子坐标信息之后是原子的初始速度信息
注:VASP 根据 POSCAR 文件确定体系的对称性。原子位置精度不够(位数太少)是一个常见错误。为更好地利用 VASP 中的对称性,强烈建议在 POSCAR 文件中指定至少 7 位有效数字的原子位置(和晶格参数,最好多一些)
其他一些例子
1
2
3
4
5
6
7
8
9
10
11
Cubic BN
3.57
0.00000000 0.50000000 0.50000000
0.50000000 0.00000000 0.50000000
0.50000000 0.50000000 0.00000000
B N
1 1
Selective dynamics
direct
0.00000000 0.00000000 0.00000000 T T F
0.25000000 0.25000000 0.25000000 F F F
MgO Fm-3m (No. 225)
1.0
2.606553 0.000000 1.504894
0.868851 2.457482 1.504894
0.000000 0.000000 3.009789
Mg O
1 1
direct
0.000000 0.000000 0.000000 Mg
0.500000 0.500000 0.500000 O
POTCAR
1
2
3
4
# POTCAR 中的 PBE 泛涵显示为 PE?
LEXCH = PE
GGA = PE
POTCAR:RCORE 代表最大截止半径,单位是波尔 bohr
PSCTR 文件:控制赝势生成文件:PSCTR
赝势目录中每个类型的泛函目录中有一个 data_base 文件,里面包含每个赝势对应元素 3 种可能结构的基态能量数据
-
赝势文件;包含计算体系中每个元素种类的赝势(元素种类的数量大于 1,只需将各元素种类的 POTCAR 文件依次连接起来即可,与 POSCAR 文件中元素种类顺序对应)
-
VRHFIN:该元素赝势的价电子排布
1
cat POTCAR.1 POTCAR.2 > POTCAR
1
2
3
4
5
grep VRHFIN POTCAR
grep TIT POTCAR
grep ENMAX POTCAR
POTCAR 示例
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
PAW_PBE Cu 22Jun2005
11.0000000000000
parameters from PSCTR are:
VRHFIN =Cu: d10 p1
LEXCH = PE
EATOM = 1390.9808 eV, 102.2342 Ry
TITEL = PAW_PBE Cu 22Jun2005
LULTRA = F use ultrasoft PP ?
IUNSCR = 1 unscreen: 0-lin 1-nonlin 2-no
RPACOR = 2.000 partial core radius
POMASS = 63.546; ZVAL = 11.000 mass and valenz
RCORE = 2.300 outmost cutoff radius
RWIGS = 2.200; RWIGS = 1.164 wigner-seitz radius (au A)
ENMAX = 295.446; ENMIN = 221.585 eV
ICORE = 3 local potential
LCOR = T correct aug charges
LPAW = T paw PP
EAUG = 586.980
DEXC = 0.000
RMAX = 2.344 core radius for proj-oper
RAUG = 1.300 factor for augmentation sphere
RDEP = 2.302 radius for radial grids
RDEPT = 1.771 core radius for aug-charge
Atomic configuration
9 entries
n l j E occ.
1 0 0.50 -8850.2468 2.0000
2 0 0.50 -1062.3498 2.0000
2 1 1.50 -916.8226 6.0000
3 0 0.50 -114.6929 2.0000
3 1 1.50 -72.1325 6.0000
3 2 2.50 -5.0394 10.0000
4 0 0.50 -4.6097 1.0000
4 1 0.50 -4.0817 0.0000
4 3 2.50 -1.3606 0.0000
Description
l E TYP RCUT TYP RCUT
2 -5.0393973 23 2.200
2 10.8846608 23 2.200
0 -4.6097109 23 2.200
0 8.2520465 23 2.200
1 -2.7211652 23 2.200
1 21.7055443 23 2.200
3 2.7211652 23 2.300
Error from kinetic energy argument (eV)
NDATA = 100
STEP = 20.000 1.050
127. 125. 125. 123. 123. 121. 119. 118.
116. 114. 113. 111. 109. 106. 104. 101.
98.2 95.5 92.6 88.3 85.3 82.3 77.8 74.7
71.6 67.1 62.5 59.5 55.1 50.8 46.6 42.6
38.7 35.0 31.5 28.2 24.2 21.4 18.0 15.7
12.9 10.5 8.46 6.71 5.25 4.04 3.05 2.10
1.52 0.979 0.608 0.404 0.234 0.133 0.701E-01 0.473E-01
0.388E-01 0.367E-01 0.365E-01 0.348E-01 0.310E-01 0.255E-01 0.195E-01 0.132E-01
0.902E-02 0.584E-02 0.423E-02 0.368E-02 0.359E-02 0.353E-02 0.322E-02 0.265E-02
0.198E-02 0.138E-02 0.933E-03 0.749E-03 0.697E-03 0.691E-03 0.639E-03 0.533E-03
0.389E-03 0.272E-03 0.210E-03 0.193E-03 0.190E-03 0.172E-03 0.136E-03 0.960E-04
0.728E-04 0.670E-04 0.655E-04 0.579E-04 0.427E-04 0.316E-04 0.274E-04 0.269E-04
0.235E-04 0.179E-04 0.134E-04 0.125E-04
END of PSCTR-controll parameters
KPOINTS
设置布里渊区 k 点网格采样大小或计算能带结构时沿高对称方向的 k 点;对 k 点进行收敛性测试是许多电子最小化计算的基本任务之一。
常规 k 点网格
1
2
3
4
5
Regular k-point mesh
0 ! 0 -> determine number of k points automatically
Gamma ! generate a Gamma centered mesh
4 4 4 ! subdivisions N_1, N_2 and N_3 along the reciprocal lattice vectors
0 0 0 ! optional shift of the mesh (s_1, s_2, s_3)
- 第 1 行:注释行
- 第 2 行:设置 k 点数目,0 表示 k 点网格自动生成
- 第 3 行:k 点网格划分方式(Monkhorst-Pack 和 Gamma 方法)
- 第 4 行:3 个方向上具体的网格划分数目
- 第 5 行:
注:Monkhorst-Pack 网格的收敛速度可能快于 Γ- 中心网格;同时需注意避免使用 Monkhorst-Pack 网格破坏对称性。
对于 hcp 结构,采用 Gamma 方法
能带计算 当性质依赖于 k 矢量时,通常沿高对称性路径将属性可视化。线模式(line mode)表示在布里渊区用户定义的点之间生成 k 点。最常用的情况是分析电子带结构。
1
2
3
4
5
6
7
8
9
10
11
12
k points along high symmetry lines
40 ! number of points per line
line mode
fractional
0 0 0 Γ
0.5 0.5 0 X
0.5 0.5 0 X
0.5 0.75 0.25 W
0.5 0.75 0.25 W
0 0 0 Γ
- 第 1 行:注释行
- 第 2 行:设置 k 点数目,非 0 数字表示每条线之间划分的 k 点数目
- 第 3 行:生成 k 点方式
INCAR
注:第一个数值为默认值
VASP 计算参数设置文件
电子优化相关参数:ALGO、ENLM、NELMIN、EDIFF、ENCUT
离子优化相关参数:IBRION、POTIM、NSW、EDIFFG
态密度积分相关参数:ISMEAR、SIGMA、LORBIT
ICHARG 随机初始化电子密度,积分值为电子数
INCAR 中的参数中的 L 开头表示逻辑数 INCAR 参数名称写错,VASP 会忽略,不影响
SYSTEM
“title string”,对体系及要执行的计算进行注释;默认值为 “unknown system”。
ENCUT
平面波截断能;收敛性测试指标之一;默认值为 POTCAR 文件中最大的 ENMAX 值
PREC
计算精度
- Accurate:
- Normal:
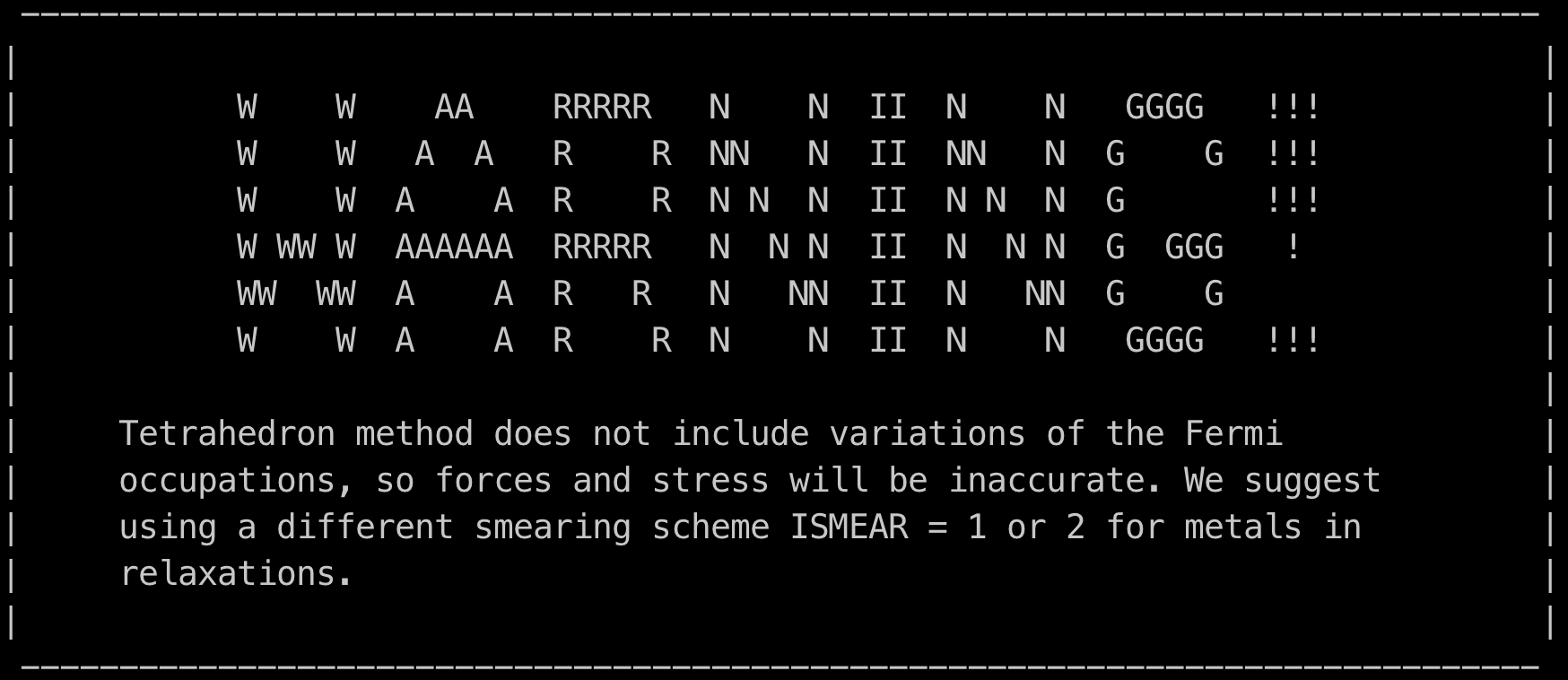
ISMEAR
轨道分数占据的展宽(平滑处理)方法
N:Methfessel-Paxton order N(默认 1)
- 0:Gaussian
- -1:Fermi
- -4:tetrahedron
- -5:Blöchl 纠正的 tetrahedron
注:
- DOS 和非常精确的总能计算(金属的非弛豫),使用 ISEMAR=-5
- 对于金属弛豫,使用 ISMEAR=1 或 ISMEAR=2 以及合适的 SIGMA 值(熵项小于 1meV/atom),合理值通常为 SIGMA=0.2(默认)
- 对于半导体或绝缘体,使用 ISEMAR=-5,胞太大或只使用 1-2 个 k 点,使用 ISMEAR=0 以及 SIGMA=0.03-0.05
tetrahedron 方法,忽略 SIGMA 参数
Tetrahedron method 需 k 点数目大于等于 4
1
2
VERY BAD NEWS! internal error in subroutine IBZKPT:
Tetrahedron method fails for NKPT<4. NKPT = 1
SIGMA
默认值:0.2 展宽宽度(单位:eV) 对于金属,默认 SIGMA=0.2,但通常 SIGMA=0.05 就能满足要求
NSW
最大离子步步数;默认值为 0。
- IBRION=0 时,必须提供 NSW 数值(ab-initio Molecular Dynamics(AIMD) 步数);
- 每个离子步会计算 Hellmann-Feynman 力和应力。
IBRION
决定离子如何更新和移动
- -1:NSW=-1 或 0,不更新;0:其他情况,执行分子动力学
- 1:RMM-DIIS 算法
- 2:conjugate gradient algorithm 共轭梯度算法
- 3:Damped molecular dynamics 算法
- 5、6:利用有限差分(finite differences)计算二阶导数、海森矩阵和声子频率
-
7、8:利用密度泛函扰动理论(density functional perturbation theory, DFPT)计算二阶导数、海森矩阵和声子频率。
- 除 0 外,其他算法都最终弛豫到局部能量最小值
- 弛豫较困难时,推荐使用 IBRION=2;在从非常糟糕的初始猜测值开始的情况下,IBRION=3 通常有用;接近能量局部最小值,推荐使用 IBRION=1
POTIM
离子弛豫步宽/AIMD 步长
ALGO
电子最小化算法/选择 GW 计算类型
- Fast:初始几步采用 blocked-Davidson(DAV) 算法,之后采用 RMM-DIIS(RMM) 算法
- Normal:blocked-Davidson 算法
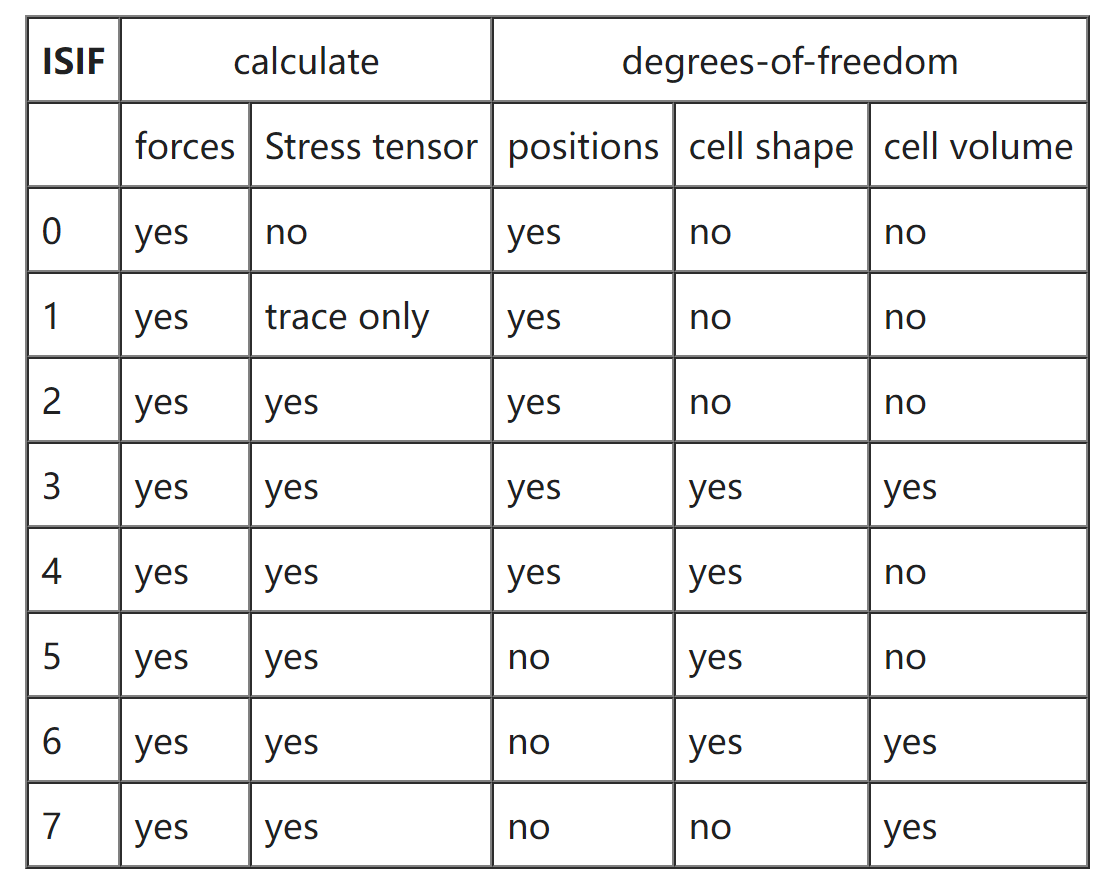
ISIF
决定在弛豫及分子动力学运行中胞的体积、形状或原子位置是否发生改变以及应力张量是否计算。
- 0:IBRION=0 时;2:其他情况
- 应力张量计算相对耗时,因此在 AIMD 中将其关掉;力总是会进行计算。
EDIFFG
离子步收敛条件
EDIFF
电子步收敛条件
NELM
每个离子步中的最大电子步数
默认值:60
NELMIN
每个离子步中的最小电子步数
LREAL
决定投影算子是在实空间还是在倒空间求值。
.FALSE.(倒空间);AUTO;.TRUE.
LCHARG
决定电荷密度是否写入 CHGCAR 和 CHG 文件中;默认值为.TRUE.。
LWAVE
决定运行结束后波函数是否写入 WAVECAR 文件中;默认值为.TRUE.。
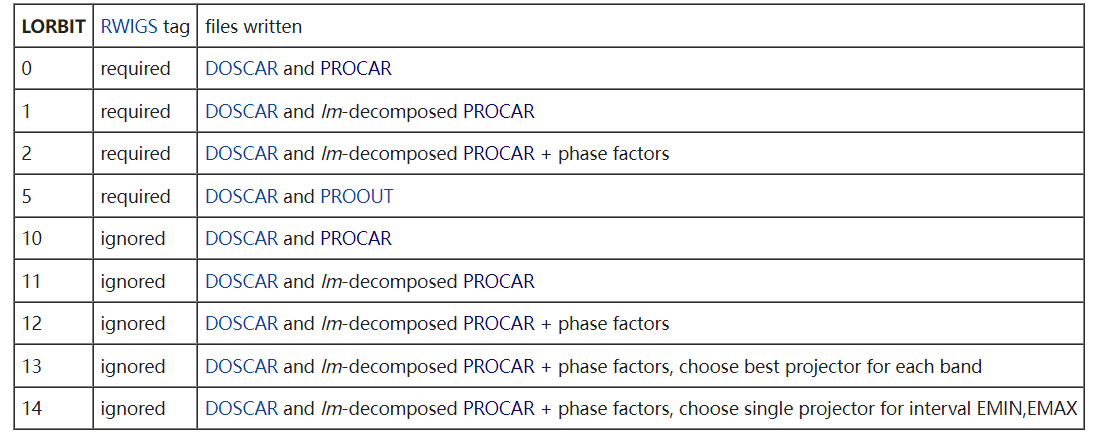
LORBIT
和适当的 RWIGS 一起,决定 PROCAR 或 PROOUT 文件是否被写入。LORBIT>=10 时,不需要 RWIGS 标签。默认值为 None。
ADDGRID
添加网格,默认值为.FALSE.;有助于降低力噪声。
SYMPREC
决定 POSCAR 文件中的位置精度,默认值为 10-5。
ISTART
初始化轨道;确定是否读取 WAVECAR 文件
默认值:1(如果 WAVECAR 文件存在)否则 0
ISYM
决定 VASP 处理对称性的方式
- 默认值:1:若 VASP 用 USPPs 运行;3:若
LHFCALC=.TRUE.;2:其他情况 -
1 2 3:对称性打开;-1 0:对称性关闭; - 与 ISYM=1 相比,ISYM=2 采用了更高效、更节省内存的电荷密度对称化方法。这尤其降低了并行版本的内存需求。
- 对于 ISYM=3,VASP 并不直接对称电荷密度。相反,电荷密度是通过对布里渊区不可还原部分 k 点处的轨道进行相关对称运算来构建的。当 LHFCALC=.TRUE 时使用这种对称方法。
- 当 ISYM=0 时,VASP 不使用对称性,但会假定 Ψk=Ψ*-k 并相应减少布里渊区的采样。这个值应该为分子动力学设置,即 IBRION=0。
-
当 ISYM=-1 时,对称性被完全关闭。
- 为什么需要对称:在 LDA 中,超胞和电荷密度的对称性总是相同的。由于在计算中使用了一组不可约对称性的 k 点,因此这种对称性被打破。为了储存正确的电荷密度和力,有必要对称这些量。
- 如果打开对称运算,则 NWRITE=3 将对称运算写入 OUTCAR 文件。
ISPIN
是否考虑自旋极化:1 - 不考虑;2 - 考虑
ICHARG
决定 VASP 如何构建初始电荷密度
默认值:ISTART=0,则 ICHARG=2;否则 ICHARG=0
- 0:从初始波函数计算电荷密度;
- 1:从 CHGCAR 文件读取;
- 2:如果 ISTART=0,取原子电荷密度的叠加;
- +10:非自洽计算(在整个电子最小化过程中电荷密度保持不变);
- 11:从 CHGCAR 文件获取(能带绘制的)本征值或给定的电荷密度的态密度(density of states (DOS))
MAGMOM
磁矩
NWRITE
决定往 OUTCAR 文件中写入多少内容
- 可选值:0-4
- 默认值:2;3 写入的内容最详细;4 只用于 debugging
- 长时间的 MD 运行,建议 NWRITE 设置为 0 或 1;短时间运行设置为 2
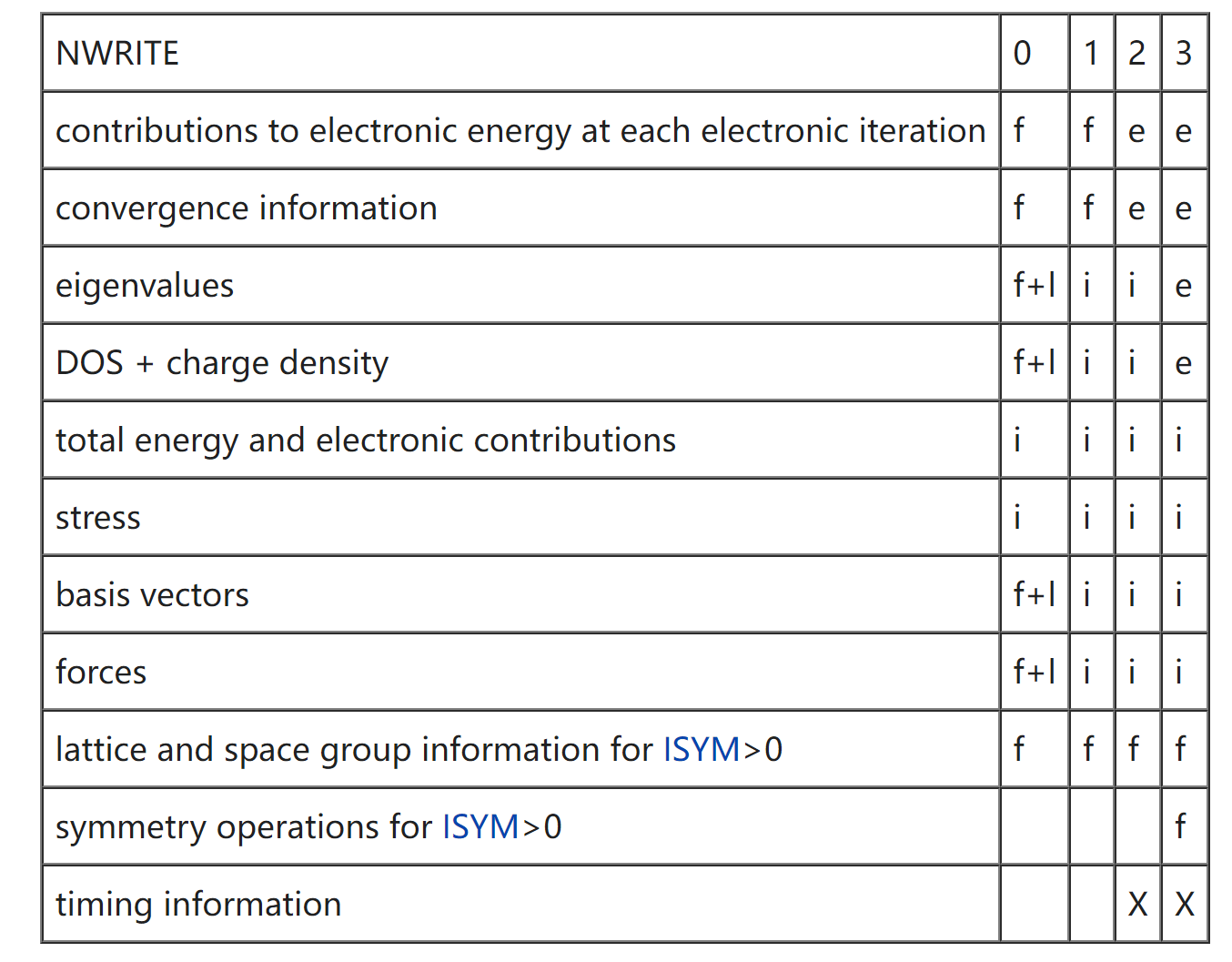
其中,f+l 表示第一步和最后一步离子步,表示 f 第一个离子步,i 表示每个离子步,e 表示每个电子步,X 表示适用时(when applicable)。
-
LAECHG = True
- 含义:用于控制是否计算电荷密度差异。
- 值:True 表示计算电荷密度差异。
-
LASPH = True
- 含义:用于控制是否考虑 LDA+U 方法中的自相互作用。
- 值:True 表示考虑自相互作用。
-
LVHAR = True
- 含义:用于控制是否计算原子的局部势能。
- 值:True 表示计算局部势能。
-
KPAR = 8
- 含义:用于并行计算中控制 k 点并行的数量。
- 值:8 表示使用 8 个处理器进行 k 点并行计算。
-
NPAR = 4
- 含义:用于控制并行计算中的分区数量。
- 值:4 表示将计算任务分成 4 个部分进行并行计算。
输出文件
输出文件结构示例
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
.
├── AECCAR0
├── AECCAR1
├── AECCAR2
├── CHG
├── CHGCAR
├── CONTCAR
├── DOSCAR
├── EIGENVAL
├── IBZKPT
├── INCAR
├── KPOINTS
├── LOCPOT
├── OSZICAR
├── OUTCAR
├── PCDAT
├── POSCAR
├── POTCAR
├── PROCAR
├── REPORT
├── slurm-2675593.out
├── sub.sh
├── vasprun.xml
├── WAVECAR
└── XDATCAR
OUTCAR
给出 VASP 计算过程的具体输出,包括:
- 输入参数的总结。
- 电子步、KS 本征值信息。
- 应力张量
- 原子受力
- 局域电荷和磁矩
- 介电性质
- 可以通过修改 INCAR 文件中的 NWRITE 标签来选择写入 OUTCAR 文件的输出量。
相关数据提取
1
reached required accuracy - stopping structural energy minimisation
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
# 构型原子数
natoms=$(sed -n '7p' POSCAR | awk '{ for(i=1; i<=NF; i++) a+=$i; print a} ')
natoms=$(grep 'NIONS' OUTCAR | tail -1 | awk '{print $12}')
# 平均原子体积
vol=$(grep 'volume of cell' OUTCAR | tail -1 | awk -v n=${natoms} '{print $5/n}')
# 平均原子能量
eng=$(grep 'free energy' OUTCAR | tail -1 | awk -v n=${natoms} '{print $5/n}')
# 磁矩 需设置 ISPIN=2
mag=$(grep 'mag=' OSZICAR | awk '{print $10}')
# 耗时
sec=$(grep 'Total CPU time used' OUTCAR | awk '{print $6}')
# 电子步能量
# 'energy without' 'free energy' 之间只有一个空格
grep 'free energy' OUTCAR
grep 'energy without entropy' OUTCAR
# 离子步能量
# 'energy without' 'free energy' 之间有两个空格
grep 'free energy' OUTCAR
grep 'energy without entropy' OUTCAR
awk 'BEGIN{i=1} /dos>/,\
/\/dos>/ \
{a[i]=$2 ; b[i]=$3 ; i=i+1} \
END{for (j=12;j<i-5;j++) print a[j],b[j]}' vasprun.xml > dos.dat
ef=`awk '/efermi/ {print $3}' vasprun.xml`
由 KPOINTS 生成的 K 点总数
1
head -n 38 IBZKPT | tail -n 35 | awk '{sum+=$4} END {print sum}'
不可约布里渊区 k 点数
1
2
3
4
grep 'irreducible k-points' OUTCAR
# output
Found 35 irreducible k-points:
能带数
1
2
3
4
grep -i 'nbands' OUTCAR
# output 也可得到不可约布里渊区 k 点数
k-points NKPTS = 35 k-points in BZ NKDIM = 35 number of bands NBANDS= 9
示例内容
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
vasp.5.4.4.18Apr17-6-g9f103f2a35 (build Nov 17 2020 17:54:46) complex
executed on LinuxIFC date 2023.09.26 17:09:42
running on 64 total cores
distrk: each k-point on 8 cores, 8 groups
distr: one band on NCORES_PER_BAND= 2 cores, 4 groups
--------------------------------------------------------------------------------------------------------
--------------------------------------- Iteration 1( 14) ---------------------------------------
POTLOK: cpu time 0.0515: real time 0.0635
SETDIJ: cpu time 0.4765: real time 0.4766
EDDAV: cpu time 0.2827: real time 0.2836
BZINTS: Fermi energy: 8.683970; 38.000000 electrons
Band energy: 134.423518; BLOECHL correction: -0.108454
DOS: cpu time 0.0149: real time 0.0149
--------------------------------------------
LOOP: cpu time 0.8256: real time 0.8387
eigenvalue-minimisations : 3224
total energy-change (2. order) :-0.3970183E-05 (-0.5334378E-05)
number of electron 37.9999994 magnetization 0.0001839
augmentation part 6.3731527 magnetization 0.0002006
Free energy of the ion-electron system (eV)
---------------------------------------------------
alpha Z PSCENC = 140.25282115
Ewald energy TEWEN = -1636.51123608
-Hartree energ DENC = -101.91636345
-exchange EXHF = 0.00000000
-V(xc)+E(xc) XCENC = -146.59065512
PAW double counting = 3226.49321421 -3016.41118853
entropy T*S EENTRO = 0.00000000
eigenvalues EBANDS = 134.42351831
atomic energy EATOM = 1329.85235582
Solvation Ediel_sol = 0.00000000
---------------------------------------------------
free energy TOTEN = -70.40753370 eV
energy without entropy = -70.40753370 energy(sigma->0) = -70.40753370
--------------------------------------------------------------------------------------------------------
FREE ENERGIE OF THE ION-ELECTRON SYSTEM (eV)
---------------------------------------------------
free energy TOTEN = -70.40753370 eV
energy without entropy= -70.40753370 energy(sigma->0) = -70.40753370
--------------------------------------------------------------------------------------------------------
POTLOK: cpu time 0.5294: real time 0.5295
--------------------------------------------------------------------------------------------------------
stress matrix after NEB project (eV)
-0.50749 -0.00000 -0.00000
0.00000 -0.50749 0.00000
0.00000 0.00000 -0.67546
FORCES: max atom, RMS 0.042143 0.029800
FORCE total and by dimension 0.084286 0.042143
Stress total and by dimension 0.985565 0.675456
LOOP+: cpu time 12.0820: real time 12.2709
4ORBIT: cpu time 0.0000: real time 0.0000
total charge
# of ion s p d tot
------------------------------------------
1 0.480 0.854 3.763 5.097
2 0.480 0.854 3.795 5.129
3 0.480 0.854 3.794 5.128
4 0.480 0.854 3.765 5.099
5 1.041 1.504 0.000 2.545
6 1.038 1.505 0.000 2.543
7 0.891 1.513 0.000 2.405
8 0.891 1.513 0.000 2.404
--------------------------------------------------
tot 5.782 9.452 15.117 30.351
magnetization (x)
# of ion s p d tot
------------------------------------------
1 -0.000 0.000 0.000 0.000
2 0.000 -0.000 -0.000 -0.000
3 -0.000 0.000 0.000 0.000
4 0.000 0.000 -0.000 -0.000
5 0.000 0.000 0.000 0.000
6 0.000 -0.000 0.000 -0.000
7 -0.000 -0.000 0.000 -0.000
8 -0.000 0.000 0.000 0.000
--------------------------------------------------
tot 0.000 0.000 0.000 0.000
BZINTS: Fermi energy: 8.683970; 38.000000 electrons
Band energy: 134.423518; BLOECHL correction: -0.108454
total amount of memory used by VASP MPI-rank0 49266. kBytes
=======================================================================
base : 30000. kBytes
nonlr-proj: 4721. kBytes
fftplans : 2354. kBytes
grid : 6243. kBytes
one-center: 124. kBytes
wavefun : 5824. kBytes
General timing and accounting informations for this job:
========================================================
Total CPU time used (sec): 16.449
User time (sec): 14.631
System time (sec): 1.818
Elapsed time (sec): 16.883
Maximum memory used (kb): 85376.
Average memory used (kb): 0.
Minor page faults: 29744
Major page faults: 6
Voluntary context switches: 2545
OSZICAR
电子步和离子步迭代的具体信息
- N: 电子结构的迭代步数,通常被称为电子步。
- E: 当前电子步的体系能量(自由能)。
- dE: 当前电子步和上一步体系能量的差值。
- d eps: 能带结构能量的变化。
- ncg: 作用于波函数的哈密顿量的求值次数。
- rms: 试用波函数(trial wavefunction)的残差的范数(即它们的近似误差)。
- rms(c): 输入输出电荷密度之差(前几步电荷密度不参与迭代)。
开启自旋计算,每个离子步后会有磁矩信息(mag=)
文件内容示例
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
N E dE d eps ncg rms rms(c)
DAV: 1 0.262963906782E+03 0.26296E+03 -0.13606E+04 2304 0.140E+03
DAV: 2 -0.147719592593E+02 -0.27774E+03 -0.26887E+03 2384 0.300E+02
DAV: 3 -0.805108877868E+02 -0.65739E+02 -0.64315E+02 3744 0.132E+02
DAV: 4 -0.839490730879E+02 -0.34382E+01 -0.34294E+01 3104 0.324E+01
DAV: 5 -0.840274618995E+02 -0.78389E-01 -0.78495E-01 3160 0.523E+00 0.415E+01
DAV: 6 -0.681149709824E+02 0.15912E+02 -0.12273E+02 3448 0.642E+01 0.127E+01
DAV: 7 -0.678322667781E+02 0.28270E+00 -0.13214E+01 3464 0.237E+01 0.918E+00
DAV: 8 -0.676127626457E+02 0.21950E+00 -0.14812E+00 3504 0.791E+00 0.140E+00
DAV: 9 -0.675766178893E+02 0.36145E-01 -0.17189E-01 3304 0.256E+00 0.398E-01
DAV: 10 -0.675781648484E+02 -0.15470E-02 -0.81316E-02 3208 0.128E+00 0.288E-01
DAV: 11 -0.675770209933E+02 0.11439E-02 -0.89472E-03 3248 0.587E-01 0.204E-01
DAV: 12 -0.675771006993E+02 -0.79706E-04 -0.23153E-03 3088 0.236E-01 0.725E-02
DAV: 13 -0.675771007310E+02 -0.31732E-07 -0.91291E-05 2928 0.667E-02
1 F= -.67577101E+02 E0= -.67577101E+02 d E =0.000000E+00 mag= -0.0000
CONTCAR
CONTCAR 中的 CONT 是继续的意思
VASP 计算结束后的构型文件
PROCAR
对于静态计算,PROCAR 文件包含每个轨道的 spd- 和位点投影波函数特征。在 VASP 中实现了多种确定投影波函数特征的方案,通常由标签 LORBIT 和 RWIGS 控制。当 LORBIT<10 时,必须在 INCAR 文件中指定 RWIGS 标签,在这种情况下,轨道将被投影到由 RWIGS 确定区域内的非零球谐波上。 当 LORBIT>=10 时,不需要 RWIGS 标签,投影将被投影到投影函数上。
LORBIT=11 时的格式
1
2
3
4
5
6
7
8
9
10
11
# of k-points: 5 # of bands: 26 # of ions: 3
k-point 1 : 0.00000000 0.00000000 0.00000000 weight = 0.06250000
band 1 # energy -17.37867948 # occ. 1.00000000
ion s py pz px dxy dyz dz2 dxz x2-y2 tot
1 0.144 0.000 0.000 0.000 0.000 0.000 0.000 0.000 0.000 0.145
2 0.291 0.000 0.006 0.000 0.000 0.000 0.000 0.000 0.000 0.298
3 0.291 0.000 0.006 0.000 0.000 0.000 0.000 0.000 0.000 0.298
tot 0.727 0.000 0.013 0.000 0.000 0.000 0.000 0.000 0.000 0.740
vasprun.xml
xml 格式的输出文件(内容和 OUTCAR 文件类似)
CHG、CHGCAR
体系的电荷密度文件,两者内容基本相同;后者文件内容还包含 PAW one-center occupancies(前者数据精度略低于后者),可用于重启 VASP 计算;前者主要用于可视化和后处理
这两个文件在每步迭代过程中都会被更新(设置 ICHARG=11 或 12 除外)
CHGCAR 文件内容:结构、电荷密度、augmentation occupancies
DOSCAR、EIGENVAL
DOSCAR:含态密度以及积分态密度
前 6 行是 header;第 6 行前两个数据表示能量范围,第三个数据表示带数,它与 INCAR 中 NEDOS 参数值是相同的,第四个表示费米能
1
2
3
4
5
6
Number of Ions (including empty spheres), Number of Ions, 0 (no partial DOS) or 1 (incl. partial DOS), NCDIJ (currently not used)
Volume of the unit cell [Angst**3], length of the basis vectors (a,b,c [m]), POTIM[s]
the initial Temperature TEBEG
'CAR'
the name of the system as given by SYSTEM in INCAR
E(max), E(min), (the energy range in which the DOS is given), NEDOS, E(fermi), 1.0000
从第 7 行开始(到第 6+NEDOS 行),每列的含义如下
1
2
3
4
# 不开启自旋
energy DOS integrated DOS
# 开启自旋
energy DOS(up) DOS(dwn) integrated DOS(up) integrated DOS(dwn)
EIGENVAL:每个 k 点的 Kohn-Sham 本征值
对于弛豫,DOSCAR 通常是没用的
PCDAT、XDATCAR
轨迹文件(AIMD 中每次输出步的离子构型)
IBZKPT
含不可约布里渊区 k 点数目、坐标及权重
与 KPOINTS 文件兼容,如果在 KPOINTS 文件中选择了自动生成 k 点网格,则会生成 IBZKPT 文件。
示例
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
Automatically generated mesh
35
Reciprocal lattice
0.00000000000000 0.00000000000000 0.00000000000000 1
0.11111111111111 0.00000000000000 0.00000000000000 8
0.22222222222222 0.00000000000000 0.00000000000000 8
0.33333333333333 0.00000000000000 0.00000000000000 8
0.44444444444444 0.00000000000000 0.00000000000000 8
0.11111111111111 0.11111111111111 0.00000000000000 6
0.22222222222222 0.11111111111111 0.00000000000000 24
0.33333333333333 0.11111111111111 0.00000000000000 24
0.44444444444444 0.11111111111111 0.00000000000000 24
-0.44444444444444 0.11111111111111 0.00000000000000 24
-0.33333333333333 0.11111111111111 0.00000000000000 24
-0.22222222222222 0.11111111111111 0.00000000000000 24
-0.11111111111111 0.11111111111111 0.00000000000000 12
0.22222222222222 0.22222222222222 0.00000000000000 6
0.33333333333333 0.22222222222222 0.00000000000000 24
0.44444444444444 0.22222222222222 0.00000000000000 24
-0.44444444444444 0.22222222222222 0.00000000000000 24
-0.33333333333333 0.22222222222222 0.00000000000000 24
-0.22222222222222 0.22222222222222 0.00000000000000 12
0.33333333333333 0.33333333333333 0.00000000000000 6
0.44444444444444 0.33333333333333 0.00000000000000 24
-0.44444444444444 0.33333333333333 0.00000000000000 24
-0.33333333333333 0.33333333333333 0.00000000000000 12
0.44444444444444 0.44444444444444 0.00000000000000 6
-0.44444444444444 0.44444444444444 0.00000000000000 12
0.33333333333333 0.22222222222222 0.11111111111111 24
0.44444444444444 0.22222222222222 0.11111111111111 48
-0.44444444444444 0.22222222222222 0.11111111111111 48
0.44444444444444 0.33333333333333 0.11111111111111 24
-0.44444444444444 0.33333333333333 0.11111111111111 48
-0.33333333333333 0.33333333333333 0.11111111111111 48
-0.22222222222222 0.33333333333333 0.11111111111111 24
-0.44444444444444 0.44444444444444 0.11111111111111 24
-0.33333333333333 0.44444444444444 0.11111111111111 24
-0.33333333333333 0.44444444444444 0.22222222222222 24
Tetrahedra
115 0.00022862368541
24 1 2 2 6
48 2 6 7 13
...
WAVECAR
波函数文件(包含波函数系数、特征值、费米权重等信息)
PROCAR
对于静态计算,PROCAR 文件含 spdand site projected wave function character of each orbital,由 LORBIT 和 RWIGS 参数控制。LORBIT>=10 时,不需要 RWIGS 参数。
删除输出文件
1
2
3
4
5
6
7
8
9
10
11
12
13
14
import os
def remove_files():
files = ['CHG', 'CHGCAR', 'POSCAR', 'INCAR', 'CONTCAR',
'DOSCAR', 'EIGENVAL', 'IBZKPT', 'KPOINTS', 'OSZICAR',
'OUTCAR', 'PCDAT', 'POTCAR', 'vasprun.xml',
'WAVECAR', 'XDATCAR', 'PROCAR',
'LOCPOT', 'AECCAR0', 'AECCAR1', 'AECCAR2']
for f in files:
try:
os.remove(f)
except OSError:
pass
示例
静态计算
WIP…
孤立原子计算
WIP…
收敛性测试
k 点和 ENCUT
弛豫计算
vaspkit 标准弛豫(SR) INCAR 示例:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
Global Parameters
ISTART = 1 (Read existing wavefunction, if there)
ISPIN = 1 (Non-Spin polarised DFT)
# ICHARG = 11 (Non-self-consistent: GGA/LDA band structures)
LREAL = .FALSE. (Projection operators: automatic)
# ENCUT = 400 (Cut-off energy for plane wave basis set, in eV)
# PREC = Accurate (Precision level: Normal or Accurate, set Accurate when perform structure lattice relaxation calculation)
LWAVE = .TRUE. (Write WAVECAR or not)
LCHARG = .TRUE. (Write CHGCAR or not)
ADDGRID= .TRUE. (Increase grid, helps GGA convergence)
# LVTOT = .TRUE. (Write total electrostatic potential into LOCPOT or not)
# LVHAR = .TRUE. (Write ionic + Hartree electrostatic potential into LOCPOT or not)
# NELECT = (No. of electrons: charged cells, be careful)
# LPLANE = .TRUE. (Real space distribution, supercells)
# NWRITE = 2 (Medium-level output)
# KPAR = 2 (Divides k-grid into separate groups)
# NGXF = 300 (FFT grid mesh density for nice charge/potential plots)
# NGYF = 300 (FFT grid mesh density for nice charge/potential plots)
# NGZF = 300 (FFT grid mesh density for nice charge/potential plots)
Electronic Relaxation
ISMEAR = 0 (Gaussian smearing, metals:1)
SIGMA = 0.05 (Smearing value in eV, metals:0.2)
NELM = 90 (Max electronic SCF steps)
NELMIN = 6 (Min electronic SCF steps)
EDIFF = 1E-08 (SCF energy convergence, in eV)
# GGA = PS (PBEsol exchange-correlation)
Ionic Relaxation
NSW = 100 (Max ionic steps)
IBRION = 2 (Algorithm: 0-MD, 1-Quasi-New, 2-CG)
ISIF = 2 (Stress/relaxation: 2-Ions, 3-Shape/Ions/V, 4-Shape/Ions)
EDIFFG = -2E-02 (Ionic convergence, eV/AA)
# ISYM = 2 (Symmetry: 0=none, 2=GGA, 3=hybrids)
态密度、能带计算
计算流程:
- 弛豫计算(初始构型很好,可忽略此步)
- 静态自洽计算
- 非自洽计算(能带:ICHARG=11,k-path)
- 非自洽计算(态密度:ICHARG=11,k 点密度变大)
Bader 电荷计算
WIP…
AIMD 计算
WIP…
功函数计算
WIP…
DFT 相关内容(梅师兄讲解):
POTCAR 中的一些参数
VRHFIN:该元素考虑的价电子(在写论文的计算 method 时会用到)
赝势种类:模守恒赝势、超软赝势(它们应用在哪些体系?)
没有磁性的构型添加自旋,计算速度会变慢(2 倍?),但并不会对计算的性质结果产生影响(可以检查添加自旋后的计算磁矩是否接近 0)
能带计算:先正常/自洽计算,生成 WAVECAR 和 CHGCAR;后进行非自洽计算
自洽与非自洽计算的区别:电子密度是否匹配;不是静态与弛豫计算的区别!
vaspkit 在 KPOINTS 文件生成的选项中,若构型是六方等对称性不是很高的结构,若网格方式选择的是 MP,最后生成的 KPOINTS 文件中的网格方式还是 Gamma(会自动纠正);推荐精度:0.03(梅);trick:每个方向上的 k 点数与其对应的晶格常数的乘积 ka 值大于 30 或 33.33,为推荐 k 点密度;每个方向上的 ka 尽可能保持相同或接近;0.03 对应的 K 点密度是 1/0.03=33.33。
(SR:标准弛豫,只弛豫原子位置;LR:点阵弛豫,全弛豫)
Γ点:原点,每个构型都有一个原点
ALGO 参数:控制电子步迭代的算法,会在 OSZICAR 中看到 DAV、RMM 等不同的算法
NELM 最大设置:300 步(梅)
NELMIN:最小电子步步数;在 AIMD 中,每个离子步中的电子步可能会很少(1-2 步),需对其进行最小步数限制(可能计算结果更准确)
对于金属体系,引入 ISMEAR 和 SIGMA 展宽后,会引入虚假温度,使得 OUTCAR 中的 T*S 项不为零,其值小于 0.001eV 时,为较好的 SIGMA 值(对于金属,ISMEAR=1 或 2,SIGMA=0.2(默认值);对于半导体,ISMEAR=-5,SIGMA=0.05)
DFT+U:计算能带
一个原子的构型也可以计算弹性常数
网络版的 vasp.5.4.4 没什么问题(VASP3 个版本都能编译成功)
VASP 相关脚本
- 检查 OUTCAR 文件中 T·S 项的数值是否小于 0.001eV,以检查 SIGMA 值是否设置合理:VASP_script/sigma.sh at master · tamaswells/VASP_script · GitHub
基于分子动力学模拟,可以通过对速度自关联函数(velocity autocorrelation function,VACF)进行傅里叶变换得到材料的振动态密度(vibrational density of states, VDOS)。VACF 是根据动力学模拟出来的轨迹文件和速度文件,求算系统在某一时刻的速度与另一时刻速度的关联程度的函数,直接看 VACF 并不能很直观的得到一些信息,而 VDOS 直接对应实验红外光谱,可以直观的对高温或高压下的振动变化情况等进行分析。